With a doubling time of 20-40 minutes depending on the culture conditions, E. coli is an organism that makes it very convenient to produce large amounts of DNA in a short amount of time (overnight). This is important because if a change to a single DNA molecule has been made, it's necessary to amplify the change and preserve it. Large amounts of DNA make it possible to sequence the DNA to verify the changes. The DNA can also be stored and then used again whenever the protein the DNA codes for is needed. So from the time a new piece of DNA is received (which might code for an interesting peptide, for example) which needs to be amplified, to the time it can be sent off for sequencing, is about a week.
Isolate plasmid DNA and transform competent cells.
Plasmid DNA is short circular double stranded DNA that generally plays a role in nature of transferring genetic traits from organism to organism. They are the "vectors" of genetic engineering and have been designed to make DNA manipulation easy. They have one or more restriction sites where they can be cut open with an enzyme, making it possible to insert new DNA at the position of the cut. The new, changed DNA is called recombinant DNA and is amplified in the growing cell. Since plasmid DNA has a much lower molecular weight than genomic DNA, it is easy to isolate it from the cell in relatively pure form. One of the gratifying things about DNA is you can see it! So if you follow the "old" miniprep procedure and precipitate out the plasmid DNA with ethanol, there is a small pellet of material you can see with the naked eye--which transforms cells easily. This is in contrast to using miniprep kits, where all you get is 50-100 ul of liquid which just looks like water. (Note, however, the miniprep kit DNA is of better quality for sequencing or for more enzyme cutting, since the kits have been optimized to give pure DNA). In order to amplify the DNA (or to make proteins coded by the DNA), it must first be used to transform E. coli which has been made competent. This involves a series of steps in which the cell walls of E. coli are made more permeable to the plasmid DNA using calcium ions and a process known as heat shock, wherein the cells + plasmids mixture is briefly heated from 0 to 42 C. In the process, some cells are transformed into a new genotype, which also sometimes results in a visible phenotype when the cells are streaked out on agar plates.
A plasmid almost always has a gene for an enzyme which is responsible for the degradation of an antibiotic. This makes it possible for the bacterium harboring the plasmid to grow in the presence of the antibiotic. Without the plasmid (and the enzyme the DNA on the plasmid codes for), the antibiotic oftentimes acts by disrupting the bacterium's ability to make cell walls, which is lethal for the cell. If the antibiotic is degraded, cell wall synthesis proceeds normally, but only for those cells which are actively making plasmid and antibiotic destroying enzyme. This means that any cells that make it into the culture which are not transformed do not survive and divide. It is interesting how low the probability is for a competent cell to acquire antibiotic immunity--just a few chances in a million. But of course once a transformed cell forms a colony, then all the cells in the colony have immunity (and are genetically identical) and can be further cultured in the presence of antibiotic.
Design short pieces of DNA.
None of this would be possible if we did not understand how information in DNA is converted into real things in living cells. Look up the Central Dogma of molecular biology if you want the whole picture. For now, focus on the idea that just three nucleotides in a row code for one of 20 amino acids, as delineated in a codon table. Since there are 4 nucleotides (ATGC), this means there are 4^3=64 possible combinations--there is some redundancy because some amino acids have as many as 6 three-nucleotide combinations which code for them. It turns out all 64 combinations are used, but only 61 are for amino acids. Three are reserved to signal a "stop" in the process of polypeptide synthesis. The three-nucleotide code is called a codon after it has been transcribed into RNA, since in the process of protein synthesis (translation) the actual codons are carried by RNA molecules. At right we see a 40 nucleotide piece of double stranded DNA and the bit of protein that it codes for above it. The actual amino acids are rendered in their one-letter abbreviations and the details are not important for now. But notice that GAG codes for E (glutamic acid) and CAG codes for Q (glutamine), while in this case the stop signal is TAA. This piece of short peptide is attached to a larger protein called GFP in the experiments carried out so far. The GFP part of the picture is implied in the "..." at the beginning of the peptide.
Visualize the Green Fluorescent Protein (GFP).
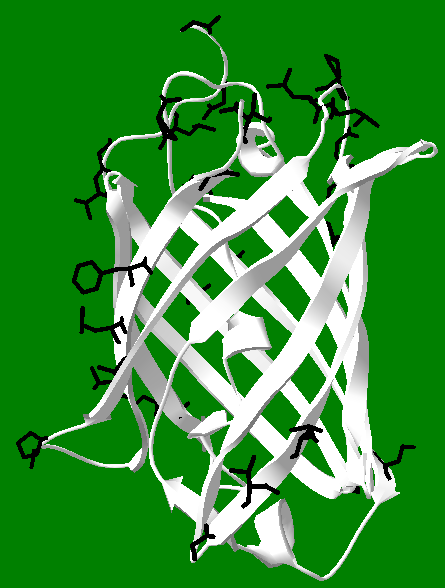
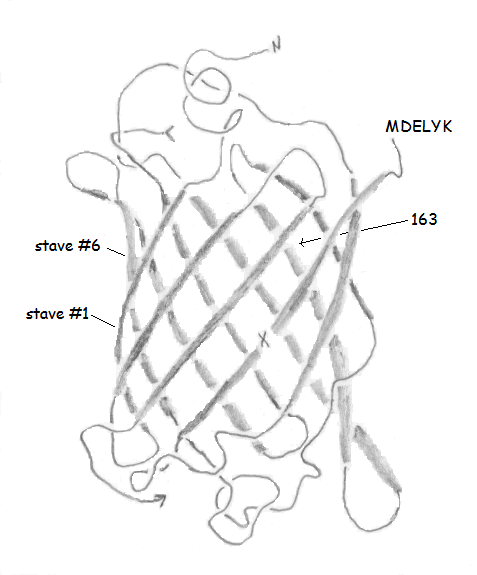
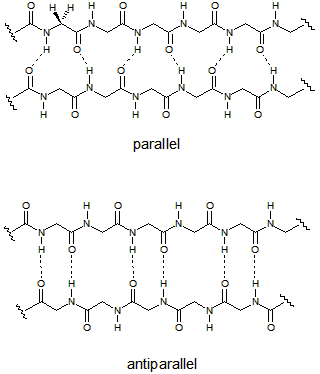
Once a string of amino acids has been attached together in sequence (and it is not a random sequence), the resulting polypeptide is in a position to fold into an active protein. There are thousands (if not millions) of different proteins, all of which play a role in cellular function. Many of them serve as enzymes, which are biological catalysts. Catalysts speed up reactions--which is crucial to the smooth functioning of most (if not all) biochemical processes. There are many other functions for proteins. We will focus on an unusual class of proteins known as fluorescent proteins (FPs), a member of which is GFP. Notice in the drawing of GFP above the 11 strands twist around a central core (the core is left empty for clarity). This is called a beta-barrel, because the individual strands take on a common form of protein called a beta sheet, and they bond to each other to form a barrel-like structure. The bonds are not depicted in the picture of GFP, but the drawings at right show the pattern of hydrogen bonding in parallel and antiparallel beta sheets. The amino acids here are glycine, with hydrogen atoms drawn on the first glycine pointing both forward and back. One of the hard parts of protein science is showing and understanding all the interactions that take place in a given protein. So for GFP, the barrel is shown to emphasize the barrel, but the bonds are not shown because they would make the image confusing. Note also the identity of the amino acids is completely absent in the drawing, again to help make it more understandable. But bear in mind the only thing that gives GFP its unique properties is the identity and sequence of the amino acids in its polypeptide chain. If you start at the "N" and follow the chain (the arrow at the bottom connects to the missing core, which would connect to the "<" if it were present), you eventually end up at "MDELYK." This final amino acid sequence is present in the wild-type protein, and gets replaced by designed tail peptides in the work described here. The image is a 3D representation of the linear chain of amino acids, and it is clear it takes a "tortuous" path. It turns out for 11 strands (or barrel staves), not all the beta interactions can be antiparallel. In fact, the bonding between staves 1 and 6 is parallel, and all the other interactions are antiparallel, such as the bonding between stave 1 and stave 2, and between staves 2 and 3.

Isolate genetically engineered proteins.
The amino acids that make up the primary sequences of all proteins can be divided evenly into two different types: polar and non-polar. Polar amino acids tend to be attracted to water, which means their side chains have water loving groups on them, like acids, bases, and alcohols. They are sometimes represented as "P" amino acids. Non-polar amino acids have the opposite affinity for water, and tend to repel it, like oil. Since they are water hating, another term for non-polar is hydrophobic. They are sometimes called "H" amino acids. Each protein has its own H/P balance. Some proteins have large H regions and tend to reside in membranes (which are like oil on the inside), while others have primarily P residues on their exteriors and are very water soluble. GFP has about 21 H residues oriented towards its exterior, shown in black in the figure at right, and many more P residues than this also oriented toward the exterior, but which are not displayed. This H/P balance makes the protein overall water soluble. These facts can be exploited in its purification using hydrophobic interaction chromatography (HIC). Under very high salt concentrations, the H portions of the protein tend to aggregate together, forming dimers and higher oligomers, and they will also interact strongly with the hydrophobic part of the HIC column. This means that GFP "sticks" to the column when loaded onto it using high salt concentrations, while other less hydrophobic proteins do not, and tend to wash off the column faster than GFP. Once the contaminating proteins have been removed, the salt concentration is lowered, making GFP dissolve to a greater extent in solution, and it moves down the column, where it can be collected as highly fluorescent fractions in relatively pure form. One can see in the figure that the black hydrophobic groups are very unevenly distributed on the surface of GFP. Possibly, hydrophobic side chains which are close together act in coordination with each other to enhance the overall binding effects of the H groups. But it is an open question if the pattern of H groups contributes to the affinity of GFP for HIC, or in the ability of GFP to form dimers and higher oligomers. This picture also illustrates the power of Deep View, which was used to generate it. After obtaining the pdb file, it is easy to select amino acids of interest and highlight them. Note this image shows the core intact, which is partially visible as a helix on the interior of the protein. The core includes the amino acids out of which the chromophore is made, but the chromophore itself is not shown.
View and measure the green fluorescence.
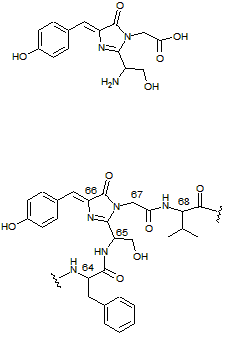
One of the more interesting phenomena in nature is fluorescence. It is a form of luminescence, which is akin to anything that "glows in the dark." In fluorescence, a chromophore absorbs light of one wavelength and emits light at another wavelength. Importantly, the energy of the incoming light is always higher than the energy of the outgoing light. The compound at right (top) will appear colorless in ordinary light, which means it does not absorb visible wavelengths of light from 400-700 nm. But below 400 nm, this compound begins to absorb light strongly, which is in the ultraviolet (UV) region. In isolation, this compound would appear unremarkable when viewed with a UV blacklight. However, when it is part of the GFP molecule, exposure to UV light causes the structure to emit a beautiful green fluorescence. The reason for this is the beta barrel protects the chromophore from collisions with solvent molecules and helps to keep the molecule rigid. Both of of these factors help the molecule emit fluorescence. The GFP chromophore is shown incorporated into a few of the core amino acids. It is made from Ser65, Tyr66, and Gly67 (the amino acids are numbered in the figure). The green emission is centered around 508 nm, which is lower in energy compared to the 395 nm UV used to excite the protein. It is interesting to note that the chromophore is synthesized soon after the protein is made, using no additional biological co-factors (molecular oxygen is required for a final oxidation step). This is part of the reason GFP is so popular as a biological label, since once the gene is expressed aerobically, the protein glows green, with no additional reagents required. Genetic engineering has produced a host of different FPs by altering the chromophore, altering the cavity in which it resides, and even altering the overall connectivity of the barrel staves, resulting in different colors, folding rates, and stabilities. Fluorescence is very useful analytically because it is very sensitive. While a molecule with a simple visible light absorbing chromophore (that is, a dye) can be used analytically, a fluorescent compound in the same setting might 10-100 times more sensitive.